Neurobiologie du vieillissement
Conférencier : David F. Tang-Wai, MDCM, FRCPC, Professeur adjoint (en neurologie), Université de Toronto ; University Health Network Memory Clinic, Toronto, ON.
Le Dr David Tang-Wai a passé en revue les mécanismes de base du vieillissement humain, et a décrit en détail l’application des théories du vieillissement aux troubles neurodégénératifs, en particulier la maladie d’Alzheimer (MA). Il a examiné la neuropathologie de la MA et ses facteurs étiologiques.
Le Dr Tang-Wai, qui a salué l’intervention des conférenciers précédents, a fait remarquer que le vieillissement repose sur des mécanismes multiples, plutôt que simples, et que tous ces facteurs impliqués dans le vieillissement neurologique et ses désordres – depuis la génétique jusqu’au stress oxydatif, le dysfonctionnement mitochondrial, et l’accumulation des cellules sénescentes – sont des processus interagissant et inextricables.
Énumérant certains des principaux changements du système nerveux avec l’âge, le Dr Tang-Wai a mentionné l’importance des changements physiologiques liés au vieillissement, comme l’atrophie cérébrale, la diminution des synthèses catécholaminergique et dopaminergique, la baisse des réflexes de redressement, et la réduction du sommeil de stade 4. Ceci se manifeste sur le plan clinique par une démarche altérée/ralentie, un balancement du corps et une certaine étourderie, ainsi que par des troubles du sommeil (p. ex., de l’insomnie). Cependant, ces changements doivent être distingués des états suggestifs de processus pathologiques tels que la démence, la dépression, les troubles du mouvement comme la maladie de Parkinson, les chutes et l’apnée du sommeil, dont aucun n’est un attribut normal du processus de vieillissement.
Des études récentes confirment qu’il existe un certain déclin cognitif universel accompagnant le vieillissement, déclin qui débute au début de l’âge adulte. L’efficacité et l’aptitude mentales atteignent leur pic vers la fin de la vingtaine, puis entament un déclin.
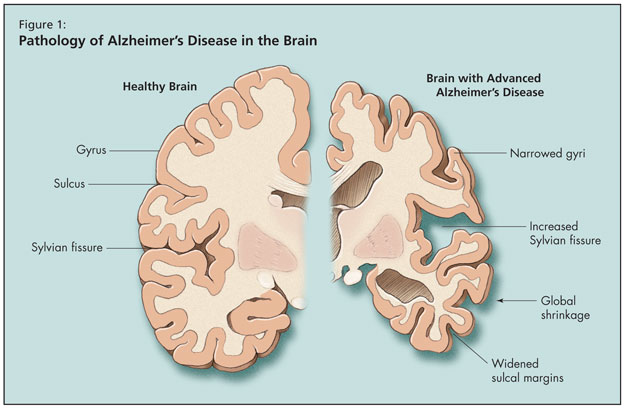
Puis le Dr Tang-Wai a fait un tour d’horizon de la MA, en notant que l’âge est le facteur de risque le plus important de la maladie, ce risque doublant après 65 ans. La maladie d’Alzheimer est la forme la plus courante de la démence de l’adulte, mais elle risque d’être bientôt dépassée par la démence mixte Alzheimer-vasculaire. Sur le plan pathologique, le cerveau subit dans la MA une atrophie globale, un élargissement des sillons corticaux, une réduction des gyri et un élargissement de la scissure de Sylvius (Figure 1).
L’évolution de la MA à l’échelle microscopique aboutit à une perte synaptique et neuronale. Finalement apparaissent les signatures pathologiques de la MA, en particulier les corps d’Hirano, les enchevêtrements neurofibrillaires, la dégénérescence granulovacuolaire et les plaques séniles.
Le Dr Tang-Wai a récapitulé la formation des enchevêtrements, qui sont le résultat de l’hyperphosphorylation des protéines tau (la protéine tau présente 6 isoformes qui s’apparient et s’agglomèrent, formant finalement des enchevêtrements), ainsi que des plaques séniles (dépôts extracellulaires d’amyloïde). Il a présenté en détail les multiples types de plaques séniles, dont la plaque diffuse du vieillissement, qui peut être observée chez la personne âgée non démente ; la plaque neuritique avec un noyau amyloïde dense ; et, avec le temps, une plaque « dégénérée » qui laisse un important noyau amyloïde. Ce processus débute dans l’hippocampe ; c’est donc la mémoire qui est touchée la première.
Discutant de la distribution des plaques et des enchevêtrements, ainsi que des stades de Braak, le Dr Tang-Wai a décrit les dérèglements initiaux de la mémoire, passant par le cortex associatif et affectant les systèmes sensoriels et moteurs au cours des derniers stades. La mention de l’hypothèse de l’amyloïde bêta (Ab) a mené à une discussion des principaux facteurs de risque de MA, notamment l’allèle de l’apolipoprotéine E (qui accélère le déclin cognitif chez la personne âgée cognitivement normale et qui est associé à une diminution de la vitesse de traitement et de l’apprentissage, ainsi qu’à une réduction du métabolisme du glucose), l’hypertension, l’obésité, l’hypercholestérolémie et le diabète. Le Dr Tang-Wai a porté une attention particulière au diabète en tant que facteur de risque dont on mesure à nouveau de plus en plus l’importance. Il a fait remarquer que l’insuline influe sur la mémoire par sa modulation de la structure et de la fonction synaptique, sa potentialisation à long terme et ses modifications des taux de neurotransmetteurs du SNC. L’insuline a des effets protecteurs sur le cerveau. Les états pathologiques d’insulinorésistance comprennent l’hyperinsulinémie, qui réduit le transport d’insuline à travers la barrière hémato-encéphalique et diminue les concentrations et l’activité de l’insuline dans le cerveau. La réduction du signal de l’insuline dans le cerveau est associée à une augmentation de la phosphorylation des protéines tau et à des concentrations plus élevées d’Ab, qui sont les signes pathologiques directs de la MA.
L’obésité et l’hypercholestérolémie ont comme effets indésirables d’augmenter l’insulinorésistance ainsi que d’augmenter les acides gras libres (AGL). De très fortes oscillations de taux d’AGL élevés entraînent une cascade d’effets indésirables. Elles inhibent les enzymes de dégradation de l’insuline, diminuant ainsi la clairance de l’Ab, et stimulant l’assemblage de la plaque amyloïde et des filaments des protéines tau. Les acides gras libres peuvent également induire une inflammation, qui stimule indirectement la formation de la plaque amyloïde et les enchevêtrements neurofibrillaires.
Parmi les principaux facteurs de risque de MA, on compte également l’hypertension de la cinquantaine, qui implique aussi une résistance à l’insuline et le risque d’un dépôt important d’Ab ; et le stress oxydatif, auquel le cerveau est particulièrement sensible, car il est largement composé de lipides facilement oxydables, il a un taux élevé de consommation d’oxygène (un quart de l’oxygène consommé va directement au cerveau), et il est dépourvu de défenses antioxydantes fortes, a encore fait remarquer le Dr Tang-Wai. Le lien entre la MA et le stress oxydatif est surtout associé à l’augmentation de l’oxydation dans le cerveau avec le vieillissement.
La dysfonction mitochondriale, ou dysfonction énergétique, joue également un rôle dans la MA. Les recherches ont montré que l’Ab peut interagir avec les mitochondries et induire une dysfonction mitochondriale. Celle-ci accélère les processus indésirables associés à la neurodégénérescence (altération de l’homéostasie calcique, génération de RLO, glycol-oxydation pouvant accélérer l’agrégation des Ab et accroître la prolifération de microglie ; mutation de l’ADN ; et altérations du traitement des protéines tau et Ab). De plus, les changements oxydatifs dans la MA sont associés aux enchevêtrements neurofibrillaires (et la théorie veut que les effets des dommages oxydatifs perturbent le neurone lui-même), ce que démontre la présence de carbonyles protéiques, de produits de la peroxydation des lipides, et des produits ultimes d’une glycation avancée.
On met de plus en plus l’accent sur le rôle de l’inflammation dans la MA par l’intermédiaire des effets synergiques de trois éléments principaux : la microglie, les astrocytes et les neurones. Les éléments inflammatoires relatifs à la neuroinflammation dans la MA comprennent la microglie et les astrocytes, qui tous deux génèrent indirectement des Ab. La microglie entoure le neurone et peut produire des radicaux libres de l’oxygène et du monoxyde d’azote, entraînant une neurodégénérescence. Les astrocytes peuvent induire une neurodégénérescence par la production de ra-dicaux libres de l’oxygène, la dégradation des Ab, et la production de cytokines et de chimiokines, favorisant encore plus l’implication de la microglie. Les neurones endommagés peuvent eux-mêmes produire des cytokines et des chimiokines, et augmenter les protéines CRP et amyloïdes P, qui activent alors le système du complément. Finalement, l’Ab peut elle-même être un facteur incitatif de la genèse des astrocytes et de l’activation microgliale. En conséquence de l’augmentation de la production de radicaux libres de l’oxygène et de la production de cytokines et de chimiokines, il y a phagocytose et dégradation de l’Ab. La protéine tau peut avoir une influence sur l’activation des systèmes du complément. En cas de régulation positive de la microglie, il peut y avoir une baisse de la dégradation de l’Ab et une baisse de l’insuline, induisant une intolérance au glucose, ce qui peut entraîner une sécrétion d’Ab. L’effet global produit boucles sur boucles de forces couplées.
Pour produire le résultat final de la pathologie de la MA, les principaux facteurs de risque et les facteurs génétiques doivent être en place. L’inflammation et le dommage oxydatif sont multisystémiques. Les processus relatifs à la pathologie de la MA présentés ici, a précisé le Dr Tang-Wai, s’appliquent à tous les processus neurodégénératifs ; ces conformations et agrégations de protéines ainsi que l’inflammation sont présentes dans les maladies de Parkinson et de Creutzfeldt-Jakob, ainsi que dans la sclérose latérale amyotrophique familiale.