Click here to view the entire report from the 4th Canadian Colloquium on Dementia
The Pros and Cons of the Amyloid Hypothesis of Alzheimer’s Disease
Speaker: Andrea LeBlanc, PhD, Professor and McGill Dawson Scholar, Department of Neurology and Neurosurgery, McGill University; Project Director, Bloomfield Centre for Research
in Aging, Lady Davis Institute for Medical Research, Sir Mortimer B. Davis Jewish General Hospital, Montreal, QC.
Researcher Dr. Andrea LeBlanc addressed evidence for and against the amyloid beta (Ab) hypothesis of Alzheimer’s pathogenesis.
Historically, Alzheimer’s disease (AD) has been characterized by its pathological signs-namely, plaques. But AD pathology encompasses more than amyloid and tau, she stated. There are degenerating neurites within those plaques that are important, as well as neurofibrillary tangles, tau, and so forth. In AD, other features play a role: synaptic loss is obvious and related to dementia but is often not discussed. Also implicated in the degenerative process are glial activation and neuronal loss. The progressive cell loss in AD is not very obvious. So, Dr. LeBlanc questioned, is it neuronal loss, neuronal atrophy and/or neuronal dysfunction causing the cognitive decline?
Arguments Marshalled in Defense of the Amyloid-Beta Hypothesis
The hypothesis holds that the Ab peptide leads to synaptic degeneration, neuronal degeneration and formation of neurofibrillary tangles and thus to the cognitive losses observed. Progressive amyloid deposition is certainly greater in AD patients, Dr. LeBlanc granted. However, some noncognitively impaired individuals have a high plaque burden. Those who defend the hypothesis suggest that they may manufacture a nontoxic form of amyloid. The correlation between amyloid deposition and impairment levels is not clear-cut; for those who doubt the amyloid hypothesis, this finding seems to disprove it.
The second argument often offered by proponents of the Ab hypothesis is that genetic mutations or polymorphisms associated with AD lead to the overproduction of Ab peptide. This is definitely true, Dr. LeBlanc stated-the amyloid precursor protein (APP), presenilin-1 and presenilin-2 genes, and related mutations have been identified by Dr. Peter St.-George Hyslop and colleagues. All of these mutations lead to increased Ab peptide levels, an increase of Ab42 versus Ab40 ratios, or an increase in Ab aggregation in the absence of total increase in Ab. These features do influence the level of Ab or its ability to aggregate. The caveat Dr. LeBlanc added is that the increases are not large-they are statistically significant increases, but their biological significance is unclear since some brains from cognitively normal individuals contain higher levels of Ab.
The third argument offered in defense of the Ab hypothesis is that research has found that increased APP expression in Down Syndrome (Trisomy 21) is associated with AD pathology. There is AD pathology in Down Syndrome individuals. The primary evidence cited for this argument concerns the case of one individual with Down Syndrome but a diploid APP, who did not show AD pathology at death. Dr. LeBlanc granted that this would appear to strongly support the amyloid hypothesis. However, Dr. LeBlanc stated, the genetic region that is duplicated in Down Syndrome contains other genes than APP that could also play a role in AD pathology.
The fourth argument often cited in defense of the hypothesis is that presenilin is the active component of gamma [g]-secretase. Presenilin-1 is part of a complex that cleaves the C-terminus of the Ab peptide from the APP. However, the g-secretase complex cleaves many other proteins. This factor’s involvement in cognitive decline is not certain, however. The function of these proteins is difficult to determine. Dr. LeBlanc suggested that these polymorphisms also alter a normal function of the protein involved in normal cognition. Work on this is underexplored compared to amyloid-seeking work, she suggested.
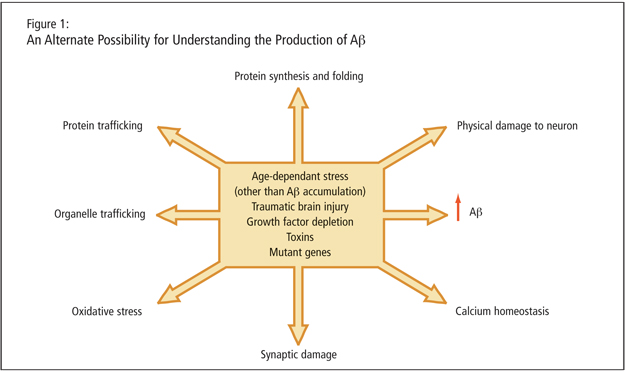
Dr. LeBlanc described age-dependent stresses, traumatic brain injury, growth factor depletion, toxins, and mutant genes as leading to increased Ab peptide. She alleged that the peptide may just be a consequence of cellular dysfunction such as protein trafficking, organelle trafficking, and oxidative stress (Figure 1). A critical experiment, she proposed, would involve altering g-secretase sites of the APP and studying the consequence. She stated that mutant genes would need to be expressed in absence of amyloid production to determine that cells are not affected in an amyloid-independent manner by the genetic mutation.
The fifth argument concerns whether Ab is a neurotoxic peptide; Dr. LeBlanc granted that this is amply demonstrated. Cytotoxicity may be associated with a very particular form of Ab, the oligomer. However, Dr. LeBlanc countered by noting that in research, cytotoxosis is induced at much higher levels than physiological concentrations. In addition, cell death is not widely evident in brains of patients with AD and is not correlated with the level of dementia experienced. Dr. LeBlanc claimed that intracellular Ab is a more compelling potential cause of AD.
The sixth argument addresses the finding that genetically engineered and transgenic animals carrying mutant AD genes generate more amyloid and develop AD pathology and cognitive impairment. These problems have been reversed by anti-amyloid immunotherapy. However, Dr. LeBlanc noted that an APP transgenic mouse with an additional mutation at the caspase C-terminal site was found to not have any cognitive impairment, despite retaining the ability to make the same amount of Ab peptide.
The final argument for the hypothesis, supported by research, is that amyloid perturbs the synaptic function. Synaptic proteins are downregulated in AD, and soluble Ab is able to dysregulate synapses. As cited by Dr. Lovestone, an Ab oligomer injected directly in the brains of mice causes LTP problems. However, Dr. LeBlanc asserted that studies are required to prove that amyloid overproduction is preceding the synaptic problems and not the other way around.
An Alternative to the Amyloid-Beta Hypothesis: Is Amyloid Production a Consequence of the Disease?
Dr. LeBlanc asked, What if amyloid is a consequence of the disease and that age-dependent stresses cause amyloid beta peptide and neuritic plaque accumulation, neurofibrillary tangles, neuritic degeneration, and synapse loss?
This has been the focus of the research conducted by Dr. LeBlanc and her colleagues over the last 14 years. Working with primary cultures of human neurons, they removed the growth factors by serum deprivation. The cultures make two to four times more Ab peptide, dependent on caspase-6. Caspase-6 is a protease that degrades other proteins when activated; it is normally present in the brain but not activated unless there is an insult. Dr. LeBlanc and colleagues designed an antibody that would recognize only the active form of caspase-6 in the brain of AD patients and found that it is associated with all neuropathological markers. To prove the enzyme would be active in AD brain, they made a second antibody directed against the tau protein cleaved by caspase-6, and found that the cleaved tau is present in pretangles, extra- and intracellular tangles, and neurophil threads. Some patients, she noted, have carpets of these threads full of tau cleaved by caspase-6. They went on to study a series of cases to investigate whether there was a correlation with cognitive impairment levels. Among their findings was that active caspase-6 was abundantly present in mild, moderate, and severe AD. They also found evidence that among noncognitively impaired subjects, there can be a large amount of neurophil threads stained for tau cleaved with caspase-6. This occurs in the absence of hyperphosphorylation of tau; Dr. LeBlanc stated that it seems to precede the other markers of tau hyperphosphorylation.
Dr. LeBlanc concluded that since there is evidence that the amyloid hypothesis does not fully account for AD pathology, research should address what could be done in addition to stopping the Ab peptide to prevent cognitive impairment.