Christine Oyugi, BSc
Managing Editor,
Geriatrics & Aging.
Edited by:
Karl Farcnik, BSc, MD, FRCPC
Psychiatrist, Division of
Geriatric Psychiatry,
University Toronto,
Part-time staff,
Toronto Western Hospital, Toronto, ON.
Contributions from:
Morris Freedman, MD, FRCPC
Director, Behavioural Neurology
Program, Baycrest Centre for Geriatric Care and Staff Scientist,
Rotman Research Institute, Toronto, ON.
Helena C. Chui, MD
Professor of Neurology,
University of Southern California
Los Angeles, Ranchos Los Amigos
National Rehabilitation Center, Downey, CA, USA.
Ian McKeith, MD, FRCPsych
Professor of Old Age Psychiatry,
Institute for Health of the Elderly
University of Newcastle Upon Tyne, UK.
- What are the clinical features of Frontotemporal and Lewy Body Dementias?
- What is the relationship between dementia and vascular disease?
- How would you differentiate among the different dementias?
- Does determining the distinction between Mild Cognitive Impairment and dementia have any clinical relevance or is it merely an academic exercise?
These are a few of the topics that were addressed by speakers during the last day of the 11th Annual Rotman Conference. This article summarizes the points presented during the last day of this conference on Tuesday, March 20th.
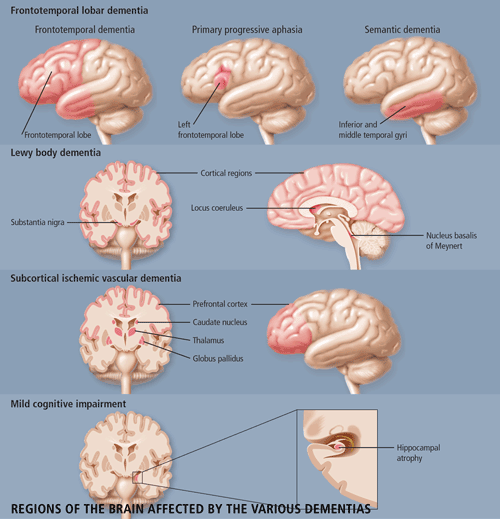
Dr. Morris Freedman, Director of the Behavioural Neurology Program at Baycrest Centre for Geriatric Care and a Staff Scientist at the Rotman Research Institute in Toronto, provided an extensive clinical review of frontotemporal dementia (FTD). The features of FTD are important for the differential diagnosis of this disease from Alzheimer disease (AD). Patients with FTD have marked personality and emotional changes that include loss of social awareness, and antisocial or disinhibited behaviour (e.g. use of rude speech, neglect of personal hygiene and grooming); they may also become easily distracted. These patients typically have poor insight and may not recognize that they have any behavioural problems. Other features include overeating, excessive smoking, oral exploration of objects and stereotypical behaviour such as wandering.
Language is a key factor in distinguishing FTD from AD. FTD patients have early preservation of language, in contrast to the situation in AD patients where language is primarily affected early in the course of the disease. As AD patients become more impaired, they develop fluent aphasia with comprehension problems. FTD patients experience a reduction in speech capacity, which may eventually lead to mutism, but their comprehension is relatively preserved. Memory loss in FTD is variable and not as severe as that with AD.
The pathophysiology of FTD involves the anterior temporal and frontal lobes. There are two forms of neuropathology--a microvacuolar form and a gliotic form. The microvacuolar form involves the general loss of neurons, microvacuolar degeneration (a spongiform-type of change), a mild astrocytic gliosis and primarily involves laminae I-III. There are no Pick cells or bodies that are seen in Pick's disease, but clinically one cannot differentiate between Pick's disease and FTD. The gliotic form of FTD represents the pathology of Pick's disease and involves all the cortical layers. There is intense astrocytic gliosis and Pick bodies may be present. There has been some debate as to whether the microvacuolar form and the Pick-type pathology represent the same or different disorders.
FTD has an earlier age of onset than does AD; the average age of onset is between 50-60 years of age. The disease duration averages 8-10 years. Although the precise cause of FTD remains unknown, there is a genetic predisposition to the disease in some patients. Fifty percent of patients with FTD have a positive family history and up to 18% have an abnormality on chromosome 17 (autosomal dominant).
There is also a relationship between FTD and motor neuron disease; some patients with FTD also develop motor neuron disease, symptoms of which can appear before or after the onset of FTD. If a diagnosis of FTD is made, it is important for physicians to be aware of the co-occurrence of motor neuron disease.
FTD patients have normal EEG results in the early phases of the disease--in fact an abnormal early EEG argues against a diagnosis of FTD. SPECT analysis shows deficits in frontal and temporal perfusion; however, this is not a diagnostic feature, as AD patients can have the same feature. Neuropsychological assessments show a marked deficit on frontal tests, with an absence of severe amnesia and perceptual 'parietal' deficits (e.g. copy to command is still good).
FTD is one of three prototypical clinical syndromes comprising the broader entity of frontotemporal lobar degeneration: Frontotemporal dementia (FTD), Primary Progressive Aphasia (PPA) and semantic dementia (SD). The difference among the three conditions is based on the location of the pathology--FTD is frontotemporal, SD involves lesions in the temporal lobes bilaterally, and PPA involves left frontotemporal pathology. SD, more so than either FTD or PPA, is easily confused with AD--similar to patients with AD, these patients have fluent speech and comprehension deficits. But unlike those with AD, these patients lose the meaning of words and objects with relatively good preservation of memory. PPA is less likely to be confused with AD because patients' speech becomes non-fluent.
Although patients show serotonergic deficits, there are currently no drugs available for the treatment of FTLD. SSRI's may improve some of the behavioural problems. As patients do not have cholinergic deficits, cholinesterase inhibitors will not help and may actually aggravate symptoms.
Dr. Ian McKeith, Professor of Psychiatry from the Institute of Health in the Elderly in Newcastle, England, updated the conference attendees on the current understanding of Lewy Body Dementia. Lewy body dementia (DLB) accounts for 15-20% of all dementias in old age, but has only been widely recognized since the mid-1990s. The clinical phenotype of Lewy Body Dementia (DLB) is related to the site, severity and amount of Lewy body pathology. DLB is characterized by the presence of Lewy bodies in the brainstem (substantia nigra and locus coeruleus), and in the subcortical (nucleus basalis of Meynert) and cortical regions of the brain. Neuronal loss and gliosis are also present in those areas. In some cases, there is an overlap between DLB, AD and Parkinson's disease (PD). As is the case in patients with both AD and PD, DLB patients have b-amyloid plaques and neurofibrillary tangles in their brains, although not in sufficient numbers to make a diagnosis of AD. The core clinical features of DLB include fluctuating cognitive impairment (seen in 80% of patients), persistent visual hallucinations (70% of patients) and Parkinsonism (75% of patients). These features are used to distinguish between DLB patients and AD patients (See Table 1). Other features which are supportive of DLB but lack specificity are: transient lack of consciousness (40%), falls and syncope (50%), systematized delusion (70%), neuroleptic sensitivity (50%), depression (50%) and REM sleep disorder (no estimates available). DLB is commonly mis-diagnosed as AD, as patients are equally impaired on both the MMSE and the Cambridge Cognitive Examination (CAMCOG). However, DLB patients perform worse on tests of attention (e.g. reaction time), visuospatial performance (e.g. clock drawing) and visual perception (e.g. fragmented letters). A Consensus guideline for the clinical and pathological diagnosis of dementia with Lewy bodies was developed in 1996. Several studies have been performed to validate these criteria and have found that the Consensus criteria for DLB performed as well in prospective studies as did those for AD and vascular dementia (VaD), with a high diagnostic sensitivity. Fluctuation is an important diagnostic indicator, reliable measures of which need to be further developed. Although specificity of the clinical diagnostic criteria is generally high, 17-78% of cases may be missed. This may be attributed to clinicians being unaware of the criteria or unfamiliar with the diagnosis. The potential contribution of neuroimaging to the differential diagnosis of DLB from other dementias remains uncertain, although relative preservation of the hippocampus and temporal lobe is found in DLB when compared with AD.
| TABLE 1 Core Clinical Feature of DLB vs. AD |
| Clinical Feature | DLB | AD |
| Fluctuating Cognitive Impairment | 80% | 60% |
| Persistent Visual Hallucinations | 70% | 15% |
| Parkinsonism (bradykinesia, rigidity, gait) | 75% | 20% |
Currently, there is no treatment that stops the progression of DLB. Much of the focus on treatment has been the management of the neuropsychiatric symptoms of the disease and the associated movement disorders. Unfortunately, 50% of patients show sensitivity to older neuroleptics including haloperidol and phenothiazines, and these patients are more likely to die than are those not treated with these drugs. However, newer antipsychotics such as olanzapine and quetiapine may be relatively safer for the management of DLB. Recent studies have shown a benefit of acetylcholinesterase inhibitors with respect to the treatment of behavioural, as well as cognitive, aspects of this disease, and it is possible that these drugs could become the treatment of choice in the future.
Dr. Helena Chui, a Professor from the University of Southern California, gave a talk on cognitive impairment due to subcortical ischemic vascular disease.
Ischemic vascular disease (IVD) is a common cause of dementia in the Western world. Similar to the situation with AD, the incidence of VaD increases with age. However, the exact incidence and prevalence of VaD is difficult to discern. The major problem remains the disagreements with regards to diagnostic criteria and their implementation. In particular, there is uncertainty regarding the following:
- The classification of patients who show both vascular and degenerative features (mixed-dementia);
- The difficulty choosing among several different clinical criteria (e.g., the Hachinski Ischemic Score);
- The use of imaging findings in defining VaD;
- The minimal level of disease severity required for a patient to be included in epidemiologic studies.
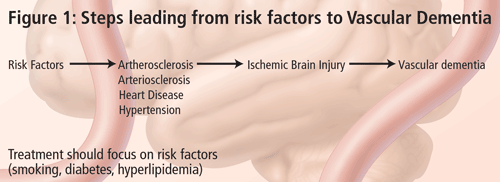
The problem in diagnosing vascular dementia lies in the causal relationship. It is not very difficult to diagnose dementia and, with the recent advancements in structural imaging, it is also not very difficult to diagnose vascular disease. The conundrum is--what is the relationship between the two and how do we know that the vascular lesion seen in imaging is causing the dementia syndrome? According to Dr. Chui, the term Vascular Dementia is too broad. VaD is not a disease, but only one possible phenotypic expression of vascular brain injury. For this reason, her talk focused on subcortical ischemic vascular dementia (SIVD). There are many types of cerebrovascular disease, leading to variable clinical and symptomatic expressions (Figure 2). There are a number of guidelines available on the effective treatment of risk factors that lead to these conditions and this should be the focus of SIVD management. The frequency of SIVD seems to vary depending on the ethnic group; it is more common in persons of Japanese or African American descent.
The small vessels affected in SIVD are within the brain parenchyma and are small penetrating arterioles approximately 100 to 600 mm in diameter. The predominant risk factors for SIVD are diabetes mellitus, hypertension, amyloid angiopathy (a subset of AD), cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). SIVD is a term that can be used for either the disease or the dementia syndrome. According to Dr. Chui, SIVD represents a more homogeneous clinical and pathological entity, which may be a more useful target for treatment, especially if the target is the cerebrovascular disease and cerebrovascular brain injury rather than its symptomatic expression.
There are two proposed underlying pathophysiologic mechanisms of how SIVD leads to ischemic brain injury--Occlusion and Hypoperfusion. Occlusion leads to small lacunar complete infarcts, cystic necrosis, and loss of all tissue elements (neurons, axons, glia, astrocytes). It leads to a more homogeneous phenotype including subcortical dementia, affective disorder such as depression, extrapyramidal signs and pure motor and sensory deficits. Hypoperfusion results from widespread narrowing of small penetrating arterioles, leading to incomplete infarction where there is a selective loss of tissue elements. For instance, in the white matter there will first be a loss of oligodendrocytes with demyelination, astrogliosis and, later, a loss of axons. Hypoperfusive ischemic brain injury has been postulated to be the cause of Binswanger syndrome, which is characterized by a combination of deep white matter changes, as well as a slowly progressive subcortical dementia, gait disturbance and urinary incontinence.
Our current understanding of how cognitive impairment relates to SIVD hinges on the lacunar hypothesis. This hypothesis states that the likelihood of dementia is related to the number, size and location of lacunar infarcts within parallel frontal subcortical loops (Prefrontal Cortex-caudate-globus pallidus-thalamus-PFC). However, recent imaging studies showed that the best correlate between dementia and SIVD was atrophy and not the volume or the number of lacunar infarcts. Clinical evaluation of SIVD should include tests of working memory, recognition memory and executive function.
The treatment of SIVD can be divided into three components. The first is primary prevention, where one tries to prevent infarction or vascular cognitive impairment by managing vascular risk factors such as hypertension and diabetes. For secondary prevention, where there is evidence of vascular brain injury--infarction, incomplete infarction or vascular cognitive impairment--the goal is to prevent the recurrence or the progression of disease. There is evidence that, even at this stage, one should continue to manage hypertension but also administer other treatments. Tertiary treatment refers to symptomatic treatment of memory and cognitive impairment. Acetylcholinesterase inhibitors are currently being studied for this purpose; currently, none have been approved for this purpose.
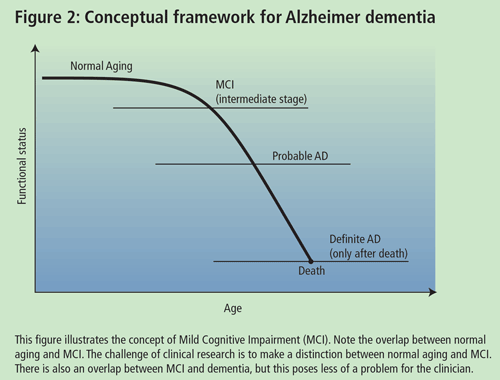
Dr. Chertkow, Associate Professor of Neurology and Neuropsychiatry at McGill University, presented a talk on high and low technological approaches to the early diagnosis of AD. According to Dr. Chertkow, the goal in trying to delineate an early mild cognitively impaired group is to identify which individuals will or will not deteriorate over finite periods of time (5-10yrs). One of the difficulties in studying individuals with Mild Cognitive Impairment (MCI) is the lack of accepted diagnostic criteria. A diagnosis of MCI can be made if patients meet the following criteria: (a) complain of defective memory; (b) normal activities of daily living; (c) normal general cognitive function; (d) abnormal memory function for age; and (e) absence of dementia. However, there are varying inclusion criteria that may overlap with the aforementioned.
Some researchers contend that patients with MCI may be an in-between group (i.e. individuals who are between normal aging and mild AD) (See Dr. Petersen's talk). The issue is to prognosticate and to define those who are going to progress from this state and those who will not. Prognosis in MCI varies depending on how you characterize your group, but severity of symptoms often predicts progression to AD. Researchers are trying to identify biological and cognitive markers that will assist the general physician in delineating MCI individuals who will progress to AD in a finite time period. The characteristic of a good marker is that it should be precise and simple, should be inexpensive, should be reliable and non-invasive and should have the ability to be validated in pathological cases. Recently, Chertkow and colleagues completed a study on mild memory loss in the elderly. The study looked at 90 individuals who passed the above criteria for MCI and followed them for 3-5 years. Over the course of the study, 51 patients deteriorated to dementia (50 of them meeting the criteria for probable AD) and 39 did not deteriorate. Initially, about 15-17% of the MCI individuals progressed to AD each year; however, even after 10 years, approximately 15% of the individuals did not have dementia and did not appear to be progressive. Therefore, there is a subgroup of MCI patients who do not progress to AD.
There were some interesting differences between those individuals that progressed to AD and those that did not. The progressing group had an older age of onset of their symptoms and performed slightly worse on the MMSE at the time of presentation. The researchers further assessed a number of clinical variables--history, risk factors for AD, physical examination--in order to identify predictors for progression of MCI to AD. The only variables useful as predictors were age, the presence of vascular disease, the number of years the individual smoked, the symptom duration and the MMSE score at initial presentation. It was suspected that some of these factors may have been explained by the same variable and a logistic regression analysis was necessary to find out which factors contribute to the prediction. When this was done, the only significant variables remaining were age at onset of memory problems and the MMSE. This predicted progression in about 67% of the MCI group. In addition, retrospectively, individuals who had lack of orientation to time in the MMSE also progressed to AD.
Hippocampal atrophy (MRI volumetrics) may also be useful as a predictor for progression. MCI patients have hippocampal volume that is intermediary between normal individuals and AD patients (who have significant shrinkage). SPECT scanning and APOE genotype did not appear be useful in predicting progression. The researchers set up an algorithm that was a combination of low-tech and high-tech measures--an approach that can be used by physicians in the future. The algorithm allows a physician to establish a score and stratifies the progression of AD in MCI individuals. In the study, individuals that scored zero on the algorithm never progressed to AD and those who scored 4 or more developed AD.
Dr. Petersen, Director of the Mayo Alzheimer's Disease Research Center, gave an update of recent clinical trials on MCI. A definitive diagnosis of AD can only be made after death through the use of neuropathological methods. For the past 15 years, there have been good criteria for probable AD and correspondence between probable AD and definite AD is about 80-90%, if the usual guidelines for diagnosis are used. Research on MCI suggests that there is a transitional point between normal aging and probable AD. The problem for a physician is how to care for a person who presents at this stage and what to tell the family.
The MCI group of patients is an important group to study because they may give us insight into normal aging. From a practical point of view, these individuals may need to be told that they have a cognitive profile that puts them at a greater risk of developing AD, although, as previously mentioned, some patients may not progress to AD. Physicians have to be very careful not to over-diagnose patients with MCI. Do people who fulfil the criteria actually progress to AD at an accelerated rate and, ultimately, can something be done to impede the development of AD in these individuals using cholinesterase inhibitors or secretase inhibitors? Dr. Petersen and colleagues obtained data from the longitudinal study on aging in Rochester, Minnesota. It should be noted that the subjects (largely Caucasian, middle income) were not necessarily representative of the general population. The cognitive function of these subjects has been followed for approximately 20 years. Usually, early in the disease, the patients are not anosagnosic but are actually aware of their memory impairment. MCI patients that meet this criteria progress to probable AD at a rate of 12% per year compared to controls that progress at a rate of about 1-2% per year. According to Dr Petersen, after 10 years, 80% of these patients progress to AD. There are qualitative features that help predict who is more likely to progress to AD and who is not. The inability of persons to benefit from cues, and hippocampal atrophy, were positive predictors of progression.
Currently, clinical trials are underway to test the use of all the second-generation cholinesterase inhibitors in MCI (Table 2) as well as vitamin E, and COX-2 inhibitors. MCI individuals, if well characterized, present a sample population that will progress to AD at a known rate and are an important target group for preventive therapy. Finding a control for this study group is difficult--age-appropriate controls could be contaminated, as they may include subjects who themselves have MCI.
| TABLE 2 Clinical Trials in MCI |
| Sponsor | Duration of Study | Endpoint | Drugs being tested |
| Alzheimer's Disease Cooperative Study (ADCS) | 3 years | Clinical probable AD | Vitamin E
Donepezil |
| Merck Frosst | 2-3 years | Clinical probable AD | Rofecoxib |
| Novartis | 2 years | Clinical probable AD | Rivastigimine |
| Janssen-Ortho | 2 years | Clinical probable AD | Galantamine |
| Pfizer | 6 months | Symptomatic improvement | Donepezil |
Most individuals with MCI will go on to develop AD. In the future, we may also determine predictive phases of other dementias, where a patient can present with slight impairment in multiple domains, or a slight impairment in a single, non-memory domain. These could be used as predictors of the development and progression of several conditions including Frontotemporal dementia, Lewy body dementia, or even primary progressive aphasia.
At least for the relationship between MCI and AD, we now have available criteria that can allow for clinical trials to determine the efficacy of intervention at this stage, possibly preventing the inevitable progression toward AD.
Dementia: Biological and Clinical Advances--Part I
Dementia: Biological and Clinical Advances--Part II
Further Readings
- Barber R, Ballard C, McKeith IG, Gholkar A, O'Brien JT, Volumetric study of dementia with Lewy bodies--A comparison with AD and vascular dementia Neurology 2000;54:1304-1309.
- Chui H. Dementia due to subcortical ischemic vascular disease. Clin Cornerstone 2001;3(4):40-51.
- Jack CR Jr, Petersen RC, Xu Y, et al. Rates of hippocampal atrophy correlate with change in clinical status in aging and AD. Neurology. 2000 Aug 22;55(4):484-89.
- Longstreth WT Jr, Manolio TA, Arnold A, Burke GL, Bryan N, Jungreis CA, et al. Clinical correlates of white matter findings on cranial magnetic resonance imaging of 3301 elderly people. The Cardiovascular Health Study. Stroke 1996 Aug;27(8):1274-82.
- McKeith IG, Ballard CG, Perry RH, Ince PG, O'Brien JT, et al. Prospective validation of consensus criteria for the diagnosis of dementia with Lewy bodies. Neurology. 2000 Mar 14;54(5):1050-8.
- Petersen RC, Stevens JC, Ganguli M, Tangalos EG, Cummings JL, DeKosky ST. Practice parameter: early detection of dementia: mild cognitive impairment (an evidence-based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology.Neurology. 2001 May 8;56(9):1133-42.
- Petersen RC. Aging, mild cognitive impairment, and Alzheimer's disease. Neurol Clin. 2000 Nov;18(4):789-806. Review.
- Petersen RC, Smith GE, Waring SC, Ivnik RJ, Kokmen E, Tangelos EG. Aging, memory, and mild cognitive impairment. Int Psychogeriatr 1997;9 Suppl 1:65-9.
- Rocca WA, Kokmen E. Frequency and distribution of vascular dementia.Alzheimer Dis Assoc Disord 1999 Oct-Dec;13 Suppl 3:S9-14.
- Shah S, Tangalos EG, Petersen RC. Mild cognitive impairment. When is it a precursor to Alzheimer's disease? Geriatrics. 2000 Sep;55(9):62, 65-8. Review.