Click here to view the entire report from the 4th Canadian Colloquium on Dementia
Curing Alzheimer’s Disease: The Case of Tau and GSK-3
Speaker: Dr. Simon Lovestone, PhD, MRCPsych, Professor, Old Age Psychiatry, MRC Centre for Neurodegeneration Research; Departments of Old Age Psychiatry and Neuroscience, King's College London, Institute of Psychiatry, London, England, UK.
The second speaker, Dr. Simon Lovestone, deemed the title of his presentation “inflammatory.” Given the complexity of AD therapeutics, can one really talk about “curing Alzheimer’s”?
Most AD treatments, Dr. Lovestone noted, are symptomatic. Disease modification therapies directed at the pathogenesis (i.e., the amyloid therapies) may or may not be curative. Dr. Lovestone explicitly defined curative treatments as those that may restore function.
Dr. Lovestone observed that most think that people with AD have symptoms because they are losing neurons. He granted the veracity of this hypothesis and that neuronal loss indeed causes symptoms. However, this notion offers little hope for people with AD, for no treatment restores neurons, short of stem-cell therapy-a therapy still in its infancy. Given this fact, and the fact that there may be no available therapy for years to come that is able to do more than slow disease progression, it would seem that there is little cause for optimism on the part of the public or researchers.
However, Dr. Lovestone rejected pessimism and suggested that treatments to restore function are within grasp. Dr. Lovestone claims that there are areas in the amyloid cascade hypothesis where restoring function is a meaningful objective-that is, from a pathogenic viewpoint, there is room to ask, could dementia symptoms result from a failure of neuronal function rather than a loss of neurons?
Tau Aggregation and Hyperphosphorylation in AD
One potential area Dr. Lovestone identified concerns tau. Neurofibrillary tangles are made of protein and tau. Tau is aggregated and phosphorylated in AD, and in neurodegenerative conditions—the tauopathies—the expression of the protein itself is altered. Dr. Lovestone advocated shifting attention from the role of the synapses and focusing instead on the protein machinery in the cell body. These proteins need to be transported to the synapse and elsewhere via axonal transport. The neurofilaments and microtubules facilitate the axonal transport needed for neuronal function. Vesicles, containing transported proteins, are held together by microtubular-associated proteins. Tau is an axonal-specific microtubular protein needed for normal neural function.
Dr. Lovestone described a hypothesis that when tau is hyperphosphorylated, or when phosphorylation is increased, tau tends to be removed. Microtubules tend to collapse and tau, under certain conditions, aggregates when not bound to microtubules. Presumably, Dr. Lovestone stated, that aggregation results in the neurofibrillary tangles of AD.
GSK-3 and Neuronal Function
Following this, Dr. Lovestone had four points to investigate: one, whether glycogen synthase kinase-3 (GSK-3) might be a tau kinase; two, whether GSK-3 might be altered in AD; three, whether there is any evidence that tau phosphorylation results in altered neuronal function; and four, whether GSK-3 might regulate neuronal function (Figure 1).
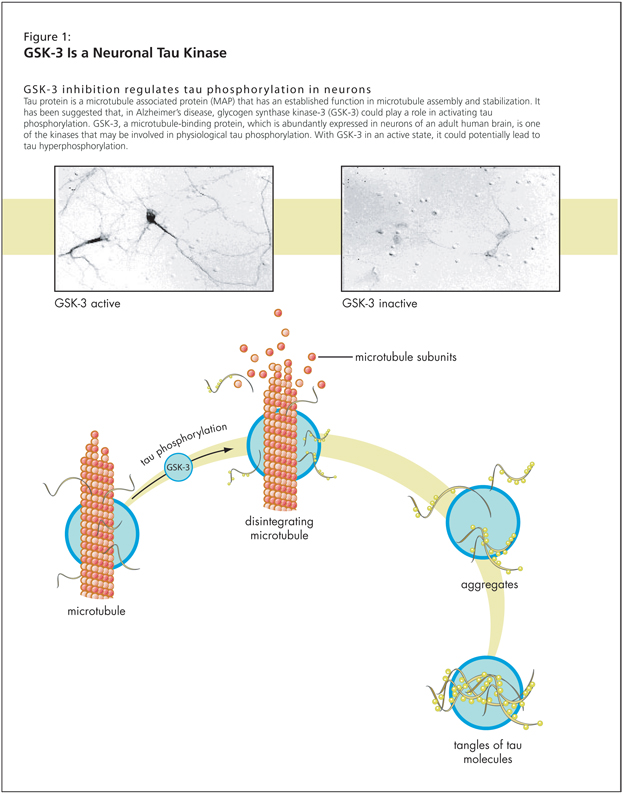
Research suggests that there is a range of kinases, including GSK-3, that will phosphorylate tau. Tau phosphorylation is switched off when the neurons are treated with a GSK inhibitor, as demonstrated in a study that used lithium. GSK-3 inhibition regulates tau phosphorylation in neurons, as a 1999 study showed.1
Regarding the second point, as to whether GSK-3 is altered in AD, Dr. Lovestone noted that it is difficult to test this hypothesis without accessing the brains of AD patients. Work by Pei et al. and Hye et al. has demonstrated that GSK-3 is increased in AD brains and that activity of GSK-3 correlates with damage. Dr. Lovestone and colleagues have investigated peripheral circulating cells; research suggests that GSK-3 is increased in white matter cells in AD.
As to whether GSK-3 protein is itself the best evidence for alteration in AD, Dr. Lovestone answers in the negative, saying evidence will likely come from understanding GSK regulation. It is inhibited by Wnt/wingless signaling and insulin signaling. Interestingly, insulin resistance is a strong risk factor for AD.
Going into the genetic evidence, Dr. Lovestone noted that a series of studies relevant to GSK, focusing on mediators of insulin signaling, have emerged from diabetes research. A diabetes study replicated with people with AD found association with a number of genes on the signaling pathway. There is further evidence from whole genome studies: according to one 2007 study, Gab2 alleles modify AD risk in APOE4 carriers, Dr. Lovestone stated. He further cited several chromosome studies showing association of mediators of insulin signaling with AD. Such studies have offered tentative evidence that regulation of GSK-3 through Wnt and/or insulin might be genetically associated with AD.
Regarding evidence that altering tau phosphorylation might be associated with altered neuronal function, Dr. Lovestone mentioned relevant drosophila studies modeling tauopathies. In such models, the addition of either human tau protein or GSK-3 affected axonal transport through clumping of the vesicles-some clumped and moved, while others were stationary. This affected neuronal function in larvae and adult flies. The phenotype effect was a massive increase of tau when they overexpressed GSK-3. The treatment of larvae with a series of GSK-3 inhibitors (i.e., lithium) reversed the phenotype in the tau-only expressing drosophila. There is a GSK-3 dependent tau phenotype in the absence of tau aggregation. In reviewing their results, they looked closely for tau aggregation, and found no evidence of it.
As to his final query-whether manipulating GSK-3 alters neuronal function-Dr. Lovestone again turned to evidence from animal studies regarding long-term potentiation (LTP), an increase in synaptic strength that follows the stimulation of the chemical synapse. In these studies of memory potentiation, they took mice with memory deficits overexpressing GSK and induced LTP. In their studies, when GSK-3 was inhibited following LTP; the memory potentiation lasted for hours. Dr. Lovestone described GSK-3 as “a gate that needs to be shut for memory to be preserved.” This may serve as early indication of the potential for treatments that restore function; however, Dr. Lovestone acknowledged, GSK-3 may prove a difficult target for pharmaceutical therapeutics to hit.
Reference
- Lovestone S, Davis DR, Webster MT, et al. Lithium reduces tau phosphorylation: effects in living cells and in neurons at therapeutic concentrations. Biol Psychiatr 1999;45:995-1003.