
Cellular Senescence
Speaker: Sharon Marr, BSc, MD, FRCP(C), MEd, McMaster University & Hamilton Health Sciences Centre, Division of Geriatrics and General Internal Medicine, Hamilton, ON.
In her address to geriatrics trainees, Dr. Sharon Marr aimed to promote a clearer understanding of the cellular changes accompanying aging, to clarify basic concepts relevant to DNA and gene expression that affect senescence, to detail cellular defense mechanisms, and to review the clinical implications of cellular senescence.
Given projected increases in the population of older adults, and the burden of age-associated disease on the health care system, a better understanding of the biology of aging is of value to clinicians. Dr. Marr distinguished between the concepts of lifespan, defined as a constant, maximum length of life for a member of a species under optimum living conditions (e.g., 120 years), and life expectancy, which is the statistically calculated estimate of the number of years of life one is expected to live at any given age.
Lifespan factors relevant to cellular changes with aging include genetic and environmental factors (e.g., impact on lifespan of caloric restriction, DNA damage/mutations, ionizing radiation, oxidative stress within or outside the cell), as well as psychological and metabolic stress, such as diabetes.
Cellular senescence (derived from the Latin senex, or old age) is fundamentally understood as a process induced by evolution into an organism’s genetic make-up. The concept encompasses all of the biological processes of a living organism as it ages. Senescence induces functional changes in cells with “full replicative potential” and in those in the post-mitotic phase.
She discussed the relevance of the Hayflick phenomenon to cellular senescence. The limit refers to the finding that human cells derived from embryonic tissues can only divide a finite number of times in culture. Dr. Hayflick and colleagues, working with human diploid fibroblasts (a cell type found in connective tissue) found that they cease to grow in vitro after a limited number of population doublings (~50 replications), a phenomenon named “replicative senescence,” which serves as a model to understand human aging.
Dr. Marr then discussed the relevance of telomere shortening. Telomeres are noncoding regions at the tips of chromosomes; this capping prevents chromosome fusions. In vertebrates, they are composed of repeated sequences of TTAGGG. With each replication our DNA starts to shorten a bit.
Telomere shortening is now considered the main causal mechanism of replicative senescence.1 Also key were James Watson’s findings that DNA polymerase could not fully synthesize the 3’ end of linear DNA. Building on the insight into incomplete replications of chromosome ends, Alexey Olovnikov observed that human somatic cells could not fully repair chromosome shortening during DNA replication, and that damage occurs each time. Olovnikov proposed that the end-replication problem would result in telomere shortening with each round of replication and that this mechanism could be the cause of replicative senescence. However, further research has shown that telomerase activity can also elongate telomeres and correct the normal telomere erosion.
Research in genetics has also provided insight into human aging and cell senescence. Accelerated aging syndromes have highlighted the role of genes, such as in Werner’s syndrome, a defect on the WRN gene, located on the short arm of the 8th chromosome. The disorder is directly caused by shorter-than-normal length telomere maintenance and impairs DNA replication.
Animal studies into telomere erosion and shortening have suggested that this shortening is correlated with aging, and that chronological age is not a predictor of life expectancy.2
In general, Dr. Marr summarized, replicative senescence is associated with progressive loss of telomeric DNA strand replication of the ends of DNA molecules and telomeres become shorter with each cell division. Certain proteins (e.g., b-Galactosidase and tumour suppressor proteins) have been found to be protective against certain cancers (p53, p21, and p16) are up-regulated. Genetic and oxidative damage also contributes to telomere shortening by direct damage to DNA.
Dr. Marr then spoke to aging’s effects on organs, which age at different rates, and then turned to age-related decrements in the immune system. The innate immune system, the first line of defense against infection, comprises neutrophils, macrophages, natural killer cells, and cytokines and chemokines. For example, neutrophils experience age-related decreases in phagocytosis, oxidative burst, and bactericidal activity, while macrophages with age lose efficiency in phagocytosis, oxidative burst, and MHC class II expression. Other key changes include promotion of a pro-inflammatory state with increased cytokines. Such a state may increase or stimulate the development of diseases such as osteoporosis, neurodegeneration, and atherosclerotic heart disease, and such a state produces degradation products that are difficult to eliminate.
In terms of altered adaptive immunity, in T-cells, thymic involution hastens progressive loss of naïve T-cells, and there is decreased ability to process new pathogens, among other deleterious effects. Key B-cell changes include reduced naïve B cells, decreased immunoglobulin generation (IgM>IgG), increased autoantibodies, and reduced antibody binding affinity
Despite reduced immunogenicity, evidence for certain vaccinations in older adults is clear. For example, the new varicella zoster vaccine found that herpes zoster was reduced by 61% and post-herpetic neuralgia by 66%.3
Regarding the implications of cell senescence on cardiovascular disease and treatment, Dr. Marr noted that coronary endothelial cells and smooth muscle cells age as we age, citing work with stains on protein deposition that were positive for b-Galactosidase (associated with senescence). Other studies investigating telomere knock out have cited an increase in cardiac myocyte proliferation, myocyte hypertrophy and heart failure, suggesting that telomere shortening with age could also contribute to cardiac failure in humans.4
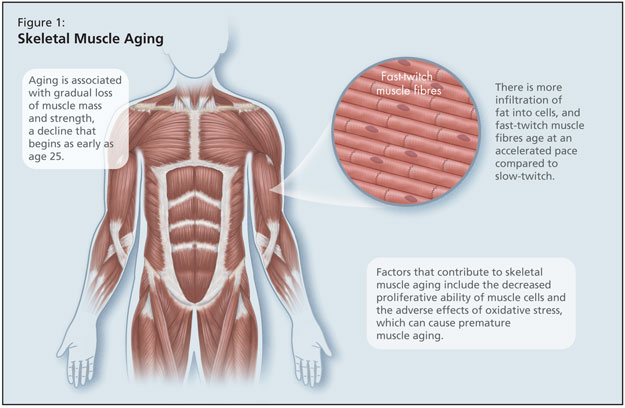
Skeletal muscle aging is also of concern, and she reminded listeners that aging is associated with gradual loss of muscle mass and strength, a decline that begins as early as age 25 (Figure 1). There is more infiltration of fat into cells, and fast-twitch muscle fibres age at an accelerated pace compared to slow-twitch.5 Factors that contribute to skeletal muscle aging include the decreased proliferative ability of muscle cells and the adverse effects of oxidative stress, which can cause premature muscle aging. In addition, there are well-known effects of aging bone, with decreased bone formation and decreased cartilage maintenance and repair.
Closing with a consideration of the clinical implications of cellular aging, Dr. Marr cited increased risk of infection, and increased burden of illness with poorer immunogenicity; however, hope lies in new research into preventing many forms of late-life cancer (e.g., via repressing ectopic telomerase or inhibition of telomerase), and the increasingly recognized importance of avoiding environmental stresses and promoting good lifestyle habits.
References:
-
Shawi M, Autexier C. Telomerase, senescence and ageing. Mech Age Devel 2008;129:3–10.
-
Bize P, Criscuolo F, Metcalfe NB, et al. Telomere dynamics rather than age predict life expectancy in the wild. Proc Biol Sci 2009;276:1679–83.
-
Oxman MN, Levin MJ, Johnson GR, et al. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med 2005;352:2271–84.
-
Leri A, Franco S, Zacheo A, et al. Ablation of telomerase and telomere loss leads to cardiac dilatation and heart failure associated with p53 upregulation. EMBO J 2003;22:131–9.
-
Lexell J. Human aging, muscle mass, and fiber type composition. J Gerontol A Biol Sci Med Sci 1995;50 Spec No:11–6.