
Neurobiology of Aging
Speaker: David F. Tang-Wai, MDCM, FRCPC, Assistant Professor (Neurology), University of Toronto; University Health Network Memory Clinic, Toronto, ON.
Dr. David Tang-Wai presented a review of the basic mechanisms of human aging and offered a detailed application of the theories of aging to neurodegenerative disorders, foremost Alzheimer’s disease (AD). He discussed the neuropathology of AD and its causative factors.
Dr. Tang-Wai, acknowledging the previous speakers, noted that aging comprises multiple rather than single mechanisms, and all of those factors concerned in neurological aging and its disorders—from genetics to oxidative stress to mitochondrial dysfunction and accumulation of senescent cells—are interacting and inextricable processes.
Enumerating some of the key changes to the nervous system with age, Dr. Tang-Wai mentioned the import of age-related physiological change, such as brain atrophy, decreased catecholaminergic and dopaminergic synthesis, decreased “righting” reflexes, and decreased stage 4 sleep. Clinically these manifest in altered/slowed gait, body sway and forgetfulness, along with sleep disturbances (e.g., insomnia). However, these changes should be distinguished from conditions suggestive of disease processes, such as dementia, depression, movement disorders such as Parkinson’s, falls, and sleep apnea, none of which are normal accompaniments to the aging process.
Recent studies confirm that some cognitive decline is universal with advancing age, a decline that begins in early adulthood. Mental efficiency and capacity peak by the late 20s and then decline.
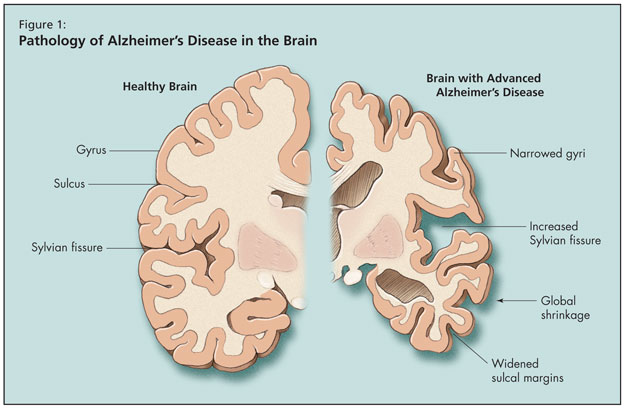
Moving on to an overview of AD, Dr. Tang-Wai noted that age is the biggest risk factor for the disease, the risk for which doubles beyond age 65. Alzheimer’s is the most common form of adult-onset dementia, but he noted that it may soon be surpassed by mixed Alzheimer-vascular dementia. What happens to the brain pathologically in AD is global shrinkage, widened sulcal margins, narrowed gyri, and increased sylvian fissure (Figure 1). As AD progresses at the microscopic level, synaptic and neuronal loss follow. Finally, pathological hallmarks of AD appear, including Hirano bodies, neurofibrillary tangles, granulovacuolar degeneration, and senile plaques.
He briefly reviewed the formation of tangles, which are the result of hyperphosphorylated tau (tau has 6 isoforms that pair up and conglomerate, ultimately forming tangles), as well as senile plaques (extracellular deposits of amyloid). He detailed multiple types of senile plaque, including diffuse plaque of aging, which may be seen in the nondemented older adult; neuritic plaque with a dense amylytic core; and with time “burned out” plaque that leaves a prominent amyloid core. This process begins in the hippocampus; thus, memory is first affected.
Discussing distribution of plaques and tangles as well as Braak staging, Dr. Tang-Wai described AD’s initial impairments in memory, progressing through the association cortex, and affecting sensory and motor systems in late stages. A mention of the amyloid beta (Ab) hypothesis prompted review of the predominant risk factors for AD, including Apolipoprotein E allele (accelerates cognitive decline in cognitively normal older adults and is associated with decreased processing speed and learning as well as reduced glucose metabolism), hypertension, obesity, hypercholesterolemia, and diabetes. Dr. Tang-Wai gave particular consideration to diabetes as a risk factor whose importance is under new and increased scrutiny. He noted that insulin influences memory by modulation of synaptic structure and function, long-term potentiation, and alterations of CNS levels of neurotransmitters. Insulin’s effects on the brain are protective. Insulin-resistant states include hyperinsulinemia, which reduces insulin transport across the blood-brain barrier and lowers insulin levels and activity in the brain. Reduced brain insulin signaling is associated with increased tau phosphorylation and higher Ab levels, which are the direct pathological signs of AD.
Obesity and hypercholesterolemia involve the adverse effects of increased insulin resistance as well as increased free fatty acids (FFA). Elevated FFA involve cascading adverse effects. They inhibit insulin-degrading enzymes, thus decreasing Ab clearance, stimulating assembly of amyloid plaque and tau filaments. Free fatty acids may also induce inflammation, which indirectly stimulates amyloid formation and neurofibrillary tangles.
Other key risk factors for AD include midlife hypertension, also involving insulin resistance and the risk of elevated Ab deposition; and oxidative stress, to which the brain is distinctly vulnerable as it is largely composed of easily oxidized lipids, has a high oxygen consumption rate (one-quarter of the oxygen consumed goes directly to the brain), and lacks strong antioxidant defenses, Dr. Tang-Wai noted. The link between AD and oxidative stress relates primarily to the increase in oxidation in the brain with aging.
Mitochondrial dysfunction, or energy dysfunction, is also implicated in AD. Research shows that Ab can interact with mitochondria and cause mitochondrial dysfunction. Mitochondrial dysfunction accelerates adverse processes associated with neurodegeneration (altered calcium homeostasis, ROS generation, glycol-oxidation, which can accelerate Ab aggregation and enhance proliferation of microglia; DNA mutation; and alterations of tau and Ab processing). Further, the oxidative changes in AD are associated with neurofibrillary tangles (and it has been theorized that effects of oxidative damage affect the neuron itself) as supported by the evidence of the presence of protein carbonyls, products of lipid peroxidation, and advanced glycation end products.
Focus is increasingly trained on the role of inflammation in AD through the synergistic effects among three major elements: microglia, astrocytes, and neurons. Inflammatory components related to AD neuroinflammation include microglia and astrocytes, both of which indirectly generate Ab. Microglia surround the neuron, and can produce reactive oxygen species and nitric oxide, leading to neurodegeneration. Astrocytes can produce neurodegeneration through production of reactive oxygen species, degradation of Ab, and production of cytokines and chemokines leading to more involvement of microglia. Damaged neurons themselves can also produce cytokines and chemokines, and increase CRP and amyloid P, which then activate the complement system. Finally, Ab itself can be an inciting factor for the genesis of astrocyte and microglial activation. Because there is increased production of reactive oxygen species and production of cytokines and chemokines, the result is phagocytosis and degradation of Ab. The tau protein can influence activation of complement systems. Microglia if upregulated can lead to decreases in Ab degradation and decreased insulin, leading to glucose intolerance, which can lead to Ab secretion. The sum effect is loops upon loops of compounding forces.
In order to produce the end result of AD pathology, key risk factors and genetic factors must be in place. Inflammation and oxidative injury are multisystemic. The processes he detailed in connection with AD pathology, Dr. Tang-Wai observed, apply to all neurodegenerative processes; these protein conformations/aggregations and inflammation are present in Parkinson’s disease and Creutzfeldt Jakob, as well as familial amyotrophic lateral sclerosis.